Research
 |
 |
 |
 |
 |
 |
Summary
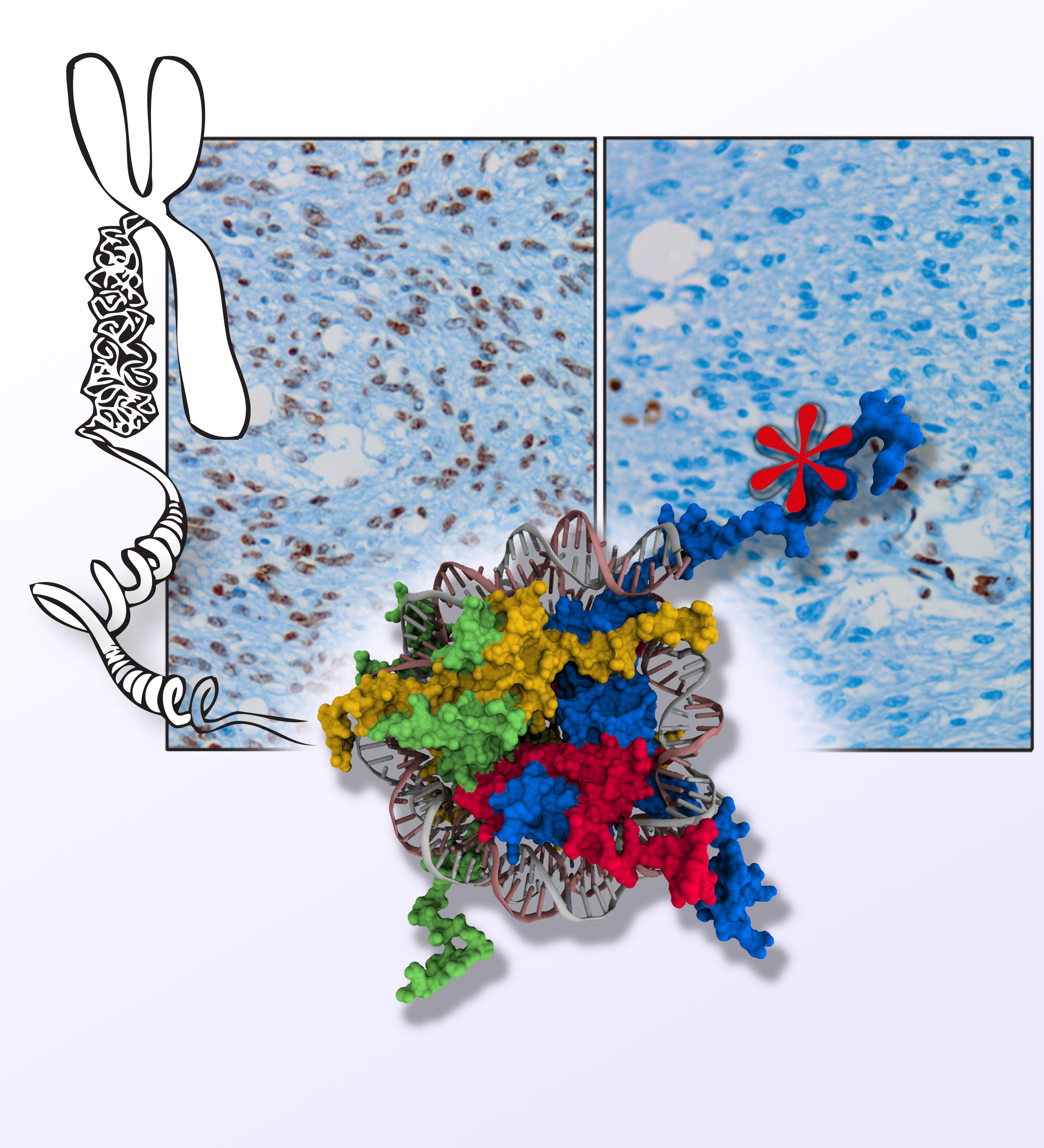
Although every gene exists within every cell in the human body, only a small percentage of genes are activated in any given cell. To manage this genetic information efficiently, nature has evolved a sophisticated system that facilitates access to specific genes. Dr. Allis studies the DNA-histone protein complex called chromatin, which packages the genetic information that exists within each cell and serves as a means of gene regulation that lies outside of the DNA itself — the basis of epigenetics.
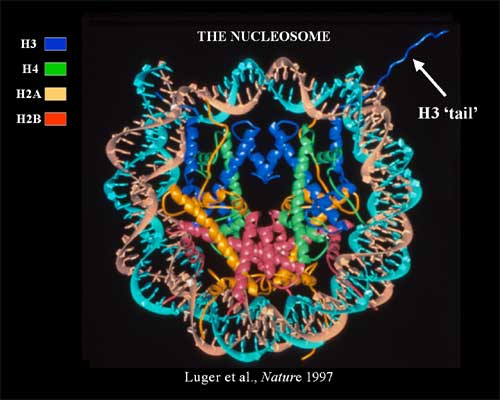
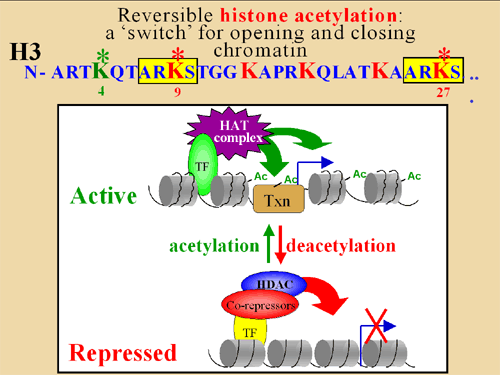
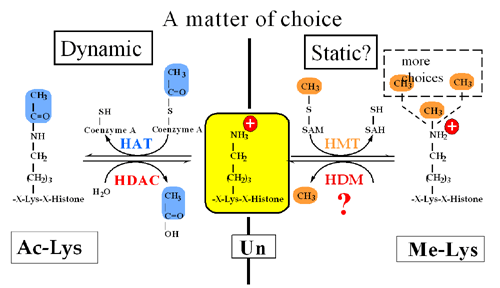
Chromatin is the physiological template of the human genome. Elaborate mechanisms have evolved to introduce meaningful variation into chromatin to alter gene expression and other important biological processes. Three major mechanisms of this are: 1) the selective use of covalent histone modifications, 2) chromatin remodeling by ATP-dependent complexes and 3) exchange of histone variants. Dr. Allis and his colleagues hypothesize that distinct patterns of covalent histone modifications form a histone or epigenetic ‘code’ that is read by effector proteins to bring about distinct downstream events. Histone proteins, their posttranslational modifications and the enzyme systems responsible for generating them are highly conserved through evolution. The Allis lab is currently investigating different histone modifications and their biological roles in a variety of mammalian cell and animal models. Through such enzymatic processes as acetylation, methylation, phosphorylation and ubiquitylation, histones are believed to function like master on/off ‘switches’ and determine whether particular genes are active or inactive. Knowing how to turn particular genes on or off could reduce the risk of certain diseases.
The frequent, high-density posttranslational modifications (PTMs) in histone proteins have lead members of the Allis laboratory to hypothesize that PTMs are located in strategic locations along the histone tail as a way for the cell to deal, reversibly, with gene activation, or in contrast, gene repression. The lab has been a front-runner in deciphering elaborate cross-talk relationships in the same histone tails (cis) or across distinct histone (trans) tails. It appears that these regulatory pathways govern chromatin function during DNA replication and repair, chromosome segregation and chromatin compaction as cells undergo apoptosis. The combinatorial read-out of distinct histone modifications that lead to the specification of unique downstream biological processes has been termed the -- the histone or epigenetic code – a widely cited and influential hypothesis.
More recently, researchers in the Allis lab recently proposed that the mammalian genome is indexed by H3 variants in a nonrandom fashion reflecting the assembly mechanisms of dedicated, personalized chaperone proteins and exchange factors that control whether genes are constitutively expressed or remain silent. The Allis lab produced the first genome-wide maps of H3.3 localization, first in mammalian embryonic stem cells and then again after the cells had differentiated to become neurons. Biochemical approaches have led them to chaperone complexes that engage H3.3 selectively, depositing it into distinct regions of the genome. One of these chaperone systems (ATRX/Daxx) targets H3.3 to telomeres and other heterochromatic loci) is mutated in a significant fraction of patients who suffer from pancreatic cancers (panNETs). Remarkably, H3 mutations (H3 lysine 27–to–methionine or H3K27M) are also highly enriched in pediatric gliomas, a disease that is clinically and molecularly distinct from its adult counterpart. We hypothesize, and have shown, that these ‘oncohistone’ mutations can alter the recruitment and activity of histone-modifying complexes, and therefore alter the epigenetic landscape and dysregulate gene expression. Given the restricted distribution of H3 mutations to pediatric gliomas, we further hypothesize that cell lineage-specific cellular context is crucial for the ability of these mutations to mediate oncogenic. In support, current findings have documented that H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates
undifferentiated sarcoma in vivo. H3K36M mutations have also been now documented in a subset of head and neck squamous cell carcinomas. New studiesidentify ENL, a translocation fusion protein of MLL (mixed lineage leukemia) as a histone acetylation reader that regulates oncogenic transcriptional programs in
acute myeloid leukemia (AML) and suggests that displacement of ENL from chromatin may be a promising epigenetic therapy alone, or in combination with, other known epigenetic inhibitors for AML. Active investigations are underway to test and extend these kinds of studies with collaborators in more clinically-relevant settings, including human patients. In unpublished studies, we investigated how recurrent, ‘hotspot’ mutations in the ENL YEATS domain -found frequently in the most common pediatric kidney cancer (Wilms’ tumor) - drive tumorigenesis.While cancer-related studies encompass much of our work in the general area of epigenetics, other areas, including neurological disorders are also under examination in the lab.
CAREER
Dr. Allis received his Ph.D. in 1978 from Indiana University and performed postdoctoral work with Martin Gorovsky at the University of Rochester. Before he joined Rockefeller in 2003 as the Joy and Jack Fishman Professor and head of laboratory, Dr. Allis held several academic positions, including ones at the Baylor College of Medicine and the University of Virginia Health System.
Dr. Allis is a member of the National Academy of Sciences and the American Academy of Arts and Sciences. Among his more recent honors are: the 2018 Lasker Basic Medical Research Award,the 2017 March of Dimes Prize, the 2016 Gruber Prize in Genetics, the 2015 Breakthrough Prize in Life Sciences and the 2014 Japan Prize.
SELECTED PUBLICATIONS
- Korb, E. et al. (2017) Synergistic translational and epigenetic disruptions in Fragile X Syndrome are improved with Brd4 inhibition. Cell170, 1209-1223 (cover)
- Wan, L. et al. (2017) ENL links histone acetylation to oncogenic gene expression in AML. Nature543, 265-269
- Papillion-Cavanagh, S. (2017) Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Genet. 49, 180-185
- Lu, C., et al. (2016) Histone H3K36 mutations impair mesenchymal differentiation and drive sarcoma development. Science352, 844-849
- Carey, B.W. et al. (2015) Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature515, 413-416
- Lewis, P.W. et al. (2013) Inhibition of PRC2 activity by gain-of-function mutations in pediatric glioblastoma. Science340, 857-861
- Elsässer, S.J. et al. (2012) Daxx envelops a histone H3.3-H4 dimer for H3.3 specific recognition. Nature 491, 560-565
- Ruthenburg, A.J. et al. (20111) Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell145, 692–706
- Goldberg, A.D. et al. (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell140, 678–691
- Wang, G.G. et al. (2009) Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature459, 847–851
- Wysocka, J. et al. (2006) A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodeling. Nature442, 86–90